|
|
|
LA CRANIOSTENOSI
La craniostenosi è definita come la prematura saldatura di una o più suture che comporta un alterato sviluppo della scatola cranica e dell�encefalo. Almeno il 20% dei casi sono causati da specifiche mutazioni di singoli geni o anomalie cromosomiche, gli altri casi sono invece dovuti a problemi di tipo meccanico o metabolico nel periodo della gravidanza (1). La precoce saldatura di una sutura determina uno spazio insufficiente per la crescita encefalica, ne consegue un incremento della pressione intracranica con conseguenze neurologiche gravi (2). L�aspetto clinico più rilevante è il cambiamento della forma del cranio, non da confondersi con altri dismorfismi cranici come le plagiocefalie posizionali. Quest�ultima è una condizione assolutamente benigna generata da un appoggio in rotazione del cranio durante la posizione supina. La pressione persistente in un lato soltanto dell�occipite determina la crescita anomala. Nella craniostenosi inoltre è spesso reperibile una rima articolare ispessita sulla sutura, significato di un rimaneggiamento dei tavolati ossei. Anche tale aspetto non è da confondersi con gli �scalini suturali� generati durante parti distocici allo scopo di diminuire la circonferenza cranica del neonato durante il passaggio nel canale del parto. L�aspetto caratteristico della sutura sinostosica è la scarsa malleabilità, poiché l�osso essendo ispessito, ha un grado di deformabilità inferiore a una sutura pervia. In tale condizione è verosimile che una palpazione esperta possa individuare precocemente lo squilibrio e consentire un rapido intervento terapeutico. Il mancato trattamento della craniostenosi può portare a deformità significative della testa, che possono diventare permanenti. Inoltre l�espansione dell�encefalo in un cranio con una crescita anomala può talvolta causare convulsioni (1,2,5).
TIPOLOGIE DI CRANIOSTENOSI
Le craniostenosi sono classificate in base alle suture interessate, e all�eventuale mutazione genetica associata.
Primaria:
La prematura fusione può avvenire a carico di una o più suture craniche:
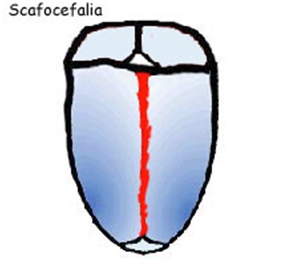
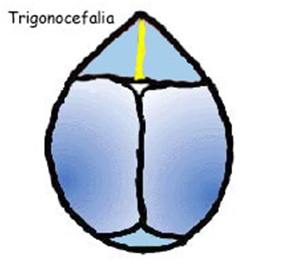
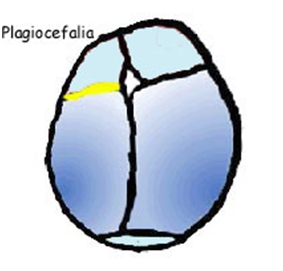
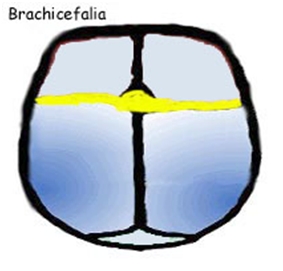
La fusione della sutura sagittale, scafocefalia, è il tipo più comune di craniostenosi, che rappresenta tra il 40% e il 55% delle cause non sindromiche. La chiusura di questa determina un cranio a forma di barca, o �cranio lungo e stretto�. La scafocefalia è più comune nei maschi che nelle femmine. La seconda forma più comune è la plagiocefalia anteriore che coinvolgere la sutura coronale. Colpisce più spesso le ragazze rispetto ai ragazzi e rappresenta tra il 20% e il 25% delle cause non sindromiche. La chiusura di entrambe le suture coronali è conosciuta come brachicefalia e determina una testa in espansione laterale. Un'altra forma è la trigonocefalia causata dalla fusione della sutura metopica, la quale ha un�incidenza altamente variabile, dal 5% al 50% di tutti i casi con una media di circa il 10%. La forma più rara è la plagiocefalia posteriore o pachicefalia, deriva dalla fusione della sutura lambdoidea e rappresenta tra il 2% e il 4% delle cause non sindromiche. Inoltre tra il 5% e il 15% delle craniostenosi ci sono forme che coinvolgono più suture. La chiusura di tutte le suture craniche è conosciuta come pansinostosi, che si presenta con un caratteristico cranio a trifoglio (1,2,3).
Sindromiche:
Circa tra il 15% e il 40% dei casi di craniostenosi sono associati a sindromi riconosciute, tra cui Apert, Carpenter, Pfeiffer, Crouzon e Chotzen (2).
EPIDEMIOLOGIA
In tutto il mondo, la craniostenosi colpisce 1 su 2000-2500 nati vivi ogni anno. Nonostante sia una condizione conosciuta da secoli, i moderni trattamenti chirurgici si sono sviluppati negli ultimi 100 anni. Il successo della chirurgia dipende dal riconoscimento precoce della craniostenosi e dalla comprensione della crescita del cranio durante l'infanzia (2).
Recenti studi hanno dimostrato un aumento del numero di pazienti cui è stata diagnosticata una craniostenosi, con un incremento relativo della percentuale di pazienti con trigonocefalia. Tuttavia, il numero di pazienti sottoposti a chirurgia è rimasto invariato. Questo può riflettere un aumento nel riconoscimento e della diagnosi delle forme meno gravi di craniostenosi (2).
EZIOPATOGENESI
La causa genetica sembra giocare un importante ruolo in alcuni tipi di craniostenosi, infatti, è spesso una caratteristica di alcune sindromi genetiche (3).
Altra causa molto comune di craniostenosi è la compressione del cranio del feto nell�utero, come può facilmente avvenire nelle gravidanze multiple, o nei casi di mioma uterino. Tuttavia disturbi come ipertiroidismo, talassemia e anemia falciforme sono associati con un aumento del rischio di craniostenosi (3).
DIAGNOSI
La tomografia computerizzata (TC) con ricostruzione tridimensionale è considerata l'indagine elettiva. La TC è in grado di rilevare chiaramente la pervietà o chiusura di ogni singola sutura. Inoltre la scansione dell�encefalo dovrebbe essere eseguita anche alla ricerca di anomalie anatomiche associate (ad esempio, ventricolomegalia e agenesia del corpo calloso) e per controllare gli spazi contenenti liquor. Una TC con venografia è richiesta in casi complessi in cui si sospetti un drenaggio venoso anomalo. Le radiografie del cranio sono di uso limitato, poiché la sensibilità nel rilevare la pervietà delle suture è significativamente inferiore alla TC. La risonanza magnetica (RMN), anche se è l�esame elettivo per indagare l�encefalo, ha una sensibilità inferiore nel visualizzare l�osso e le suture (4).
Secondo Johnson e Wilkie le craniostenosi dovrebbero essere gestite in un contesto multidisciplinare. Una valutazione completa dovrebbe includere anche quella neuropsicologica, delle componenti linguistiche, dell'udito e quella ortottica (1).
TIPI DI TRATTAMENTO
La chirurgia è il trattamento elettivo per la craniostenosi, ed ha tre obiettivi principali: correggere la deformità del cranio, prevenire la progressione e ridurre il rischio futuro d�ipertensione cranica. Questo risultato è ottenuto con l�osteotomia delle suture prematuramente fuse e la ricostruzione delle parti interessate del cranio. Il risultato è un miglioramento estetico del cranio del bambino.
C'è molta polemica nella letteratura per quanto riguarda la tempistica ottimale d�intervento chirurgico in pazienti con craniostenosi (7). Un intervento tempestivo può teoricamente prevenire disturbi neuro-cognitivi, ma, un intervento troppo precoce comporta un rischio per la vita dovuto alla perdita di sangue durante la chirurgia e può portare a una recidiva della craniostenosi. Tuttavia statisticamente gli interventi chirurgici sono eseguiti tra i sei mesi e i due anni di vita (8). E� consigliato un regolare follow-up per tutta l'infanzia, soprattutto per monitorare i sintomi di aumento della pressione intracranica, come: cefalea, alterazioni comportamentali, o il declino nel rendimento scolastico (8).
La procedura operativa per la correzione di questo disturbo dipende dal tipo di craniostenosi (interessamento di una sutura singola o di multiple) e dall�età d�insorgenza. Se il paziente presenta meno di tre mesi di età, con fusione di una sutura singola, si esegue una craniectomia endoscopica seguita da un periodo di terapia con casco. Tuttavia, se il paziente ha più di tre mesi, o ha una craniostenosi che coinvolge più suture, il percorso consigliato è una ricostruzione cranica con apertura della volta (6,9,10).
BIBLIOGRAFIA
1. Johnson D, Wilkie AO. Craniosynostosis. European Journal of Human Genetics (2011) 19, 369�376.
2. Hoey AW, Carson BS, Dorafshar AH. Craniosynostosis. Eplasty. 2012; 12: ic2.
3. Moeckel E, Mitha N. Textbook of pedistric osteopathy. Churchill Livingstone Elseviere. 2009.
4. Kirmi O, Lo SJ, Johnson D, Anslow P: Craniosynostosis: a radiological and surgical perspective. Semin Ultrasound CT MR 2009; 30: 492�512.
5. Woods RH, Ul-Haq E, Wilkie AOM et al: Reoperation for intracranial hypertension in TWIST1-confirmed Saethre-Chotzen syndrome: a 15-year review. Plast Reconstr Surg 2009; 123: 1801�1810.
6. Kapp-Simon KA, Speltz ML, Cunningham ML, Patel PK, Tomita T. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23:269�281.
7. Mehta VA, Bettegowda C, Jallo GI, Ahn ES. The evolution of surgical management for craniosynostosis. Neurosurg Focus. 2010;29(6):E5. Review.
8. Panchal J, Marsh JL, Park TS, Kaufman B, Pilgram T, Huang SH. Sagittal craniosynostosis outcome assessment for two methods and timings of intervention. Plast Reconstr Surg. 1999;103(6):1574-84.
9. Jimenez, DF, Barone CM. Endoscopic craniectomy for early surgical correction of sagittal craniosyn- ostosis. J Neurosurg. 1998;88(1):77-81.
10. Ridgway EB, Berry-Candelario J, Grondin RT, Rogers GF, Proctor MR. The management of sagit- tal synostosis using endoscopic suturectomy and postoperative helmet therapy. J Neurosurg Pediatr. 2011;7(6):620-6
|
| ||
|
Copyright 2012 Dr. Giacomo Margiacchi | Via Roma 7 - 52100 AREZZO | p.iva 01975610518
|



